
Research
Pain & Psychosocial
May 29, 2026
Immune Control of Pain and Exploration of Underlying Physiological Mechanisms - A Dive Into Theoretical Considerations to Guide the Physiotherapy Approach
Introduction
Physiotherapists might sometimes feel overwhelmed with patients’ unresolved pain, with only moderate effects of conservative treatment on patients’ complaints. Our understanding of pain and disability is largely based on the pain drivers and disability model, which has allowed a better understanding of the different contributing factors. As cognitivo-emotional drivers gained more interest in pain treatment approaches, this could have led the clinician to disregard nociceptive domains, especially in the chronic pain context. This review aims to provide physiotherapists with accessible foundational knowledge on immune control of pain to broaden clinical understanding and perspective.
Methods
This narrative review was published in the Joint Bone Spine international peer-reviewed journal.
Results
Pain classification
Nociceptive pain arises from the activation of peripheral nociceptors in response to changes in the local tissue environment. Nociceptors express various molecular sensors, including transient receptor potential (TRP) channels, G protein-coupled receptors (GPCRs), and voltage-gated sodium channels, which detect mechanical, chemical, and thermal stimuli. Once activated, they transmit electrical signals to the central nervous system.
Immune cells can contribute to the persistence of inflammatory signaling in certain chronic conditions, prolonging nociceptor sensitization. In disorders such as rheumatoid arthritis, pain may persist even when overt inflammatory activity is reduced, highlighting the complex interaction between immune control of pain and nociceptive signaling.
Neuropathic pain results from a lesion or disease of the somatosensory nervous system that alters its function. Common conditions include multiple sclerosis and diabetic neuropathy. It is often characterized by allodynia (pain elicited by normally non-painful stimuli) and hyperalgesia (exaggerated pain response to noxious stimuli). Neuropathic pain may become chronic due to persistent structural and molecular changes in affected nerves.
Nociplastic pain is defined as pain arising from altered nociceptive processing despite no clear evidence of tissue damage or somatosensory system lesion. This paper does not further detail this pain phenotype. Conditions such as fibromyalgia and some presentations of chronic low back pain are commonly associated with nociplastic pain mechanisms.
This paper highlights the contribution of the immune control of pain. While pain was historically considered a neuronal process primarily, growing evidence indicates that immune mechanisms play a key role in its modulation.
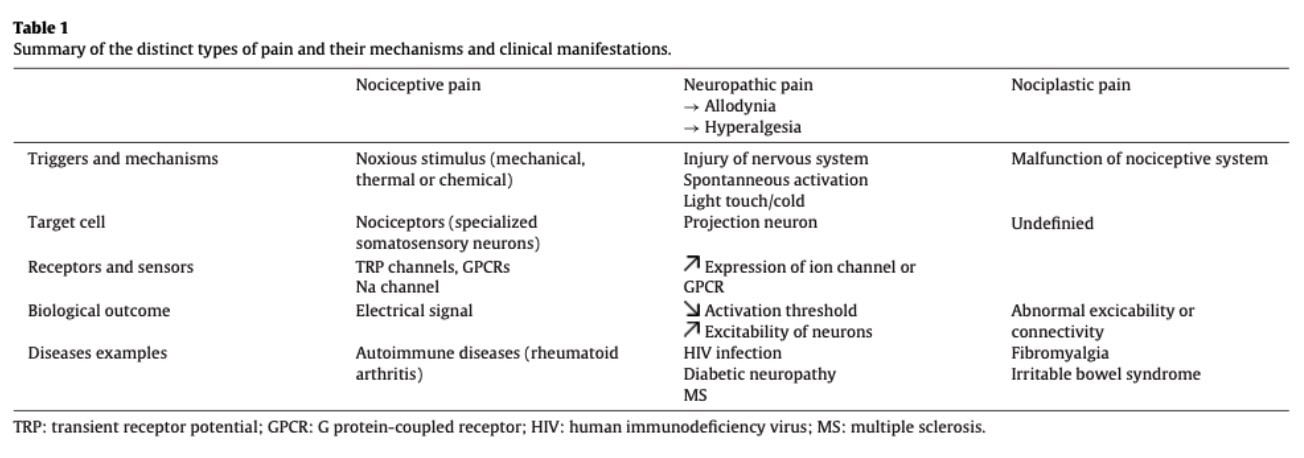
Immune cells as modulators of pain
Various immune cell types have been identified as contributors to pain mechanisms.
Macrophages are highly plastic immune cells that adopt a spectrum of activation states, often simplified as pro-inflammatory and pro-repair phenotypes (M1- and M2-like). Following tissue injury, they release mediators such as IL-1β, TNF-α, IL-6, and chemokines, which contribute to nociceptor sensitization and inflammatory pain.
During the resolution phase, macrophages shift toward a pro-resolving phenotype, reducing inflammatory signaling and promoting tissue repair. They can also release endogenous opioid peptides that activate opioid receptors on nociceptors, contributing to peripheral analgesia. In addition, macrophage-derived IL-10 helps limit inflammation and supports pain resolution.
Macrophages and nociceptors engage in bidirectional communication. Nociceptor-derived mediators such as CGRP, substance P, and chemokines, including CCL2, can modulate macrophage recruitment and activation states. This neuroimmune crosstalk can either amplify or resolve pain depending on the inflammatory context, and its dysregulation is implicated in chronic pain persistence.
Microglia are the resident immune cells of the central nervous system, often described as CNS-resident macrophage-like cells. Following nervous system injury or stress, extracellular ATP is released and activates purinergic receptors such as P2X4 and P2X7 on microglia.
This activation promotes microglial release of mediators, including brain-derived neurotrophic factor (BDNF), which acts on spinal projection neurons and modulates their excitability. This contributes to neuronal sensitization within pain pathways and plays a key role in the development and maintenance of neuropathic pain.
T cells are adaptive immune cells increasingly recognized as important regulators of pain. Different subsets fulfill distinct roles, particularly CD4+ T helper cells, which can contribute to pain by producing cytokines such as IFN-γ and IL-17, as well as cytotoxic mediators including granzyme and perforin. IL-17 has been shown to promote mechanical hyperalgesia, and its neutralization reduces pain sensitivity in animal models, highlighting its relevance in inflammatory conditions such as arthritis.
Beyond their pro-nociceptive effects, CD4+ T cells are also involved in pain modulation, as T-cell deficiency impairs endogenous analgesic mechanisms, including opioid-mediated pain control.
T cells also participate in bidirectional neuroimmune communication by expressing receptors for neuronal neuropeptides. Neuropeptides such as substance P and CGRP can influence T-cell differentiation and promote pro-inflammatory phenotypes such as Th17, thereby contributing to pain sensitization.
B-cells are antibody cells (immunoglobulin). Specific B-cells, called the Immunoglobulin G, could be implicated in neuropathic pain. An increased level of Immunoglobulin G was found in the dorsal root of mice and chronic pain patients. Depleting immunoglobulin G, on the other hand, prevented allodynia in mice. Immunoglobulin G (IgG) can contribute to nociceptive sensitization through Fcγ receptor signaling, which is expressed on nociceptive afferents. This mechanism may enhance pain sensitivity in inflammatory and autoimmune conditions, and in some experimental models, it can contribute to pain independently of overt tissue damage. As such, targeting antibody-mediated immune mechanisms may represent a promising strategy in conditions such as rheumatoid arthritis.
Neutrophils are early responders of the innate immune system and are highly inflammatory. Their role in pain is context-dependent. In acute pain, neutrophil depletion often does not affect pain sensitivity, suggesting that acute pain is primarily driven by direct nociceptor activation and early inflammatory mediators. In chronic pain models, neutrophils may infiltrate sensory ganglia, such as the dorsal root ganglia, contributing to the maintenance of pain hypersensitivity. Distinct neutrophil phenotypes are likely to have different functional effects. Clinically, patients who recover from acute low back pain show an early increase in circulating neutrophils compared to those who develop chronic pain, highlighting a potential role for early immune responses in pain resolution.
Natural Killer (NK) cells are part of the innate immune system. Their role is to detect abnormal or damaged cells and induce their elimination. Their contribution to sciatica has been identified: following axonal injury, debris accumulates within the nerve environment, and NK cells participate in its clearance, thereby promoting a healthy regenerative milieu. NK cells may also play a regulatory role by targeting hyperexcitable neurons, dysfunctional support cells, and persistent inflammatory drivers. These findings suggest that NK cells could be of interest for understanding and potentially modulating persistent pain states.
Mast cells are located in close proximity to peripheral nerve endings and contribute to pain modulation by releasing pro-inflammatory mediators such as histamine, cytokines, and proteases. These substances activate nociceptors and promote sensitization of pain pathways. Mast cells also recruit innate immune cells, thereby amplifying the inflammatory cascade and further enhancing peripheral sensitization.
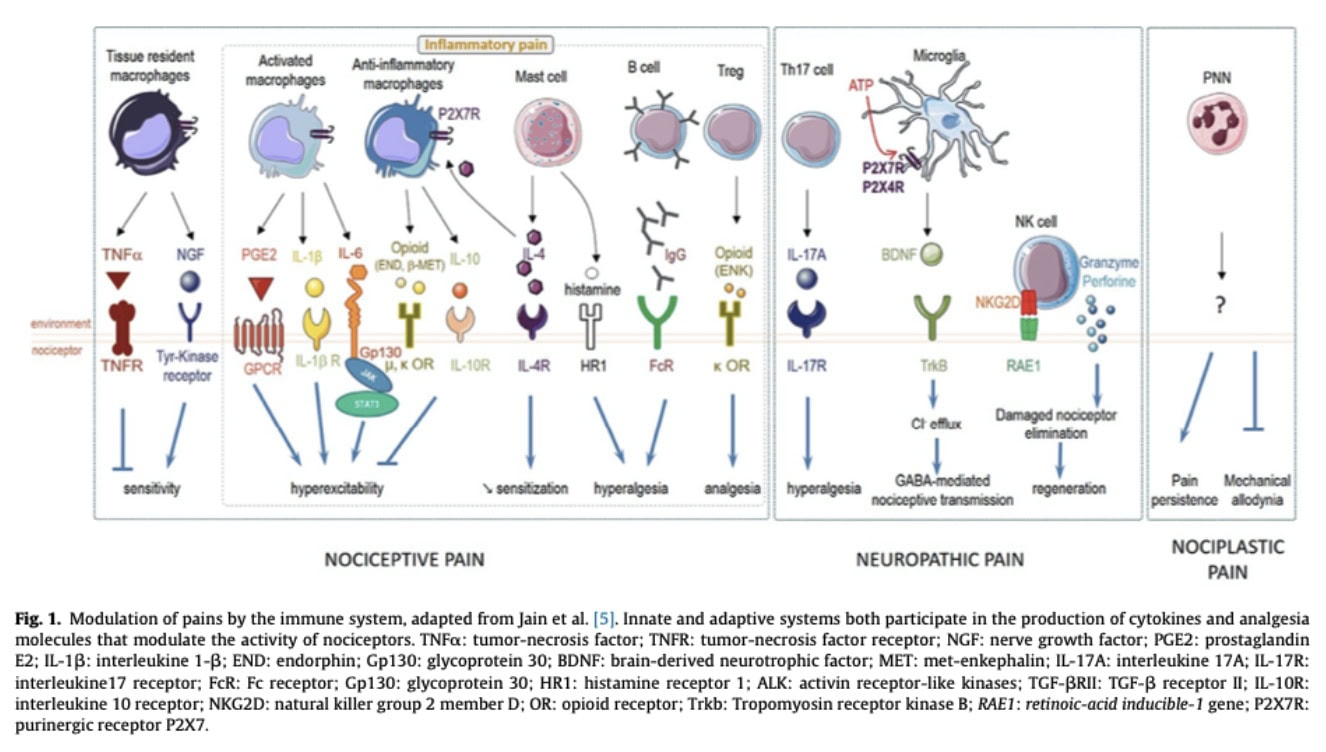
Peripheral sensitization
Peripheral sensitization refers to the increased responsiveness and reduced activation threshold of nociceptors following tissue injury or inflammation. Damaged tissue cells, resident immune cells, and recruited immune cells release inflammatory mediators such as TNF-α, IL-1β, IL-6, prostaglandins, and chemokines, which act directly or indirectly on nociceptors to increase their excitability and amplify pain signaling.
Neuroimmune pathways
Immune cells modulate nociceptor activity through the secretion of cytokines and inflammatory mediators, including IL-1β, IL-6, IL-17, IFN-γ, prostaglandin E2 (PGE2), histamine, and chemokines. Conversely, the nervous system can regulate immune activity through the release of neuropeptides such as CGRP and substance P, which influence immune cell recruitment and activation. This bidirectional communication contributes to the amplification or resolution of pain.
Central sensitization
Persistent peripheral nociceptive input may induce hyperexcitability within the central nervous system, particularly in dorsal horn neurons of the spinal cord and supraspinal pain-processing regions. This process, known as central sensitization, is associated with amplified pain perception, allodynia, widespread pain, and persistent pain even after tissue healing.
Immune contribution to central sensitization
Immune cells contribute to central sensitization through mechanisms involving microglial activation, cytokine release, and modulation of neuronal ion channels and receptors. For example, microglia detect extracellular ATP through purinergic receptors such as P2X4 and P2X7, leading to the release of mediators, including brain-derived neurotrophic factor (BDNF), which enhances neuronal excitability. Chronic pain may also alter immune function through activation of the hypothalamic-pituitary-adrenal axis and sympathetic nervous system, potentially contributing to dysregulated immune responses and impaired endogenous pain modulation.
Immunocepetion
Immunoception refers to the ability of the central nervous system to monitor and regulate immune activity. Neurons express receptors capable of detecting inflammatory signals, including cytokine receptors such as TNFR and pattern-recognition receptors such as TLR4. Through these mechanisms, the CNS can detect immune activation and adapt behaviour, metabolism, and physiological responses accordingly.
Immunoengramm
The immunengram refers to a proposed neural representation or memory trace of previous immune states, distributed between the central nervous system and peripheral immune tissues. This concept suggests that immune experiences may be encoded not only within immune cells but also within neural networks involved in neuroimmune communication. This emerging paradigm may help explain how neural activity influences immune regulation and potentially contributes to autoimmune disease processes.
Immune self-regulation and pain resolution
While many immune cells contribute to pain sensitization and nociceptive modulation, the immune system also possesses endogenous regulatory mechanisms involved in the resolution of inflammation and pain. Immune cells may therefore promote either pro-nociceptive or anti-nociceptive effects depending on the biological context, timing, and microenvironment.
Immune contribution to acute nociception
Although nociceptive pain occurs rapidly and is predominantly mediated by neuronal activation, immune-derived signals also contribute to nociceptor regulation. Molecules such as nerve growth factor (NGF), produced by immune and tissue cells, exert a tonic sensitizing effect on nociceptors by modulating their activation threshold and excitability. The absence of NGF is associated with reduced sensitivity to painful stimuli, emphasizing the contribution of immune signaling even in acute nociception. Tumor necrosis factor alpha (TNF-α) signaling is also involved in nociceptive regulation. In the absence of TNF-α signaling, nociceptors display increased sensitivity to NGF and abnormal axonal growth, resulting in enhanced pain responses.
Pro- and anti-nociceptive immune mediators
Immune cells release numerous cytokines and mediators that directly influence nociceptor excitability and pain modulation. While pro-inflammatory cytokines such as IL-1β, IL-6, TNF-α, and IL-17 promote nociceptor sensitization and inflammatory pain, anti-inflammatory cytokines, including IL-10, IL-4, and TGF-β, exert analgesic effects. IL-10 can directly act on nociceptors to reduce pain signaling, whereas IL-4 inhibits nociceptor sensitization and promotes endogenous opioid production by macrophages
Regulatory T cells and chronic pain transition
The transition from acute to chronic pain may partly depend on immune regulatory mechanisms, particularly those involving regulatory T cells (Tregs). Tregs suppress excessive inflammatory responses through the inhibition of effector immune cells and the secretion of anti-inflammatory cytokines. Experimental models of sciatic nerve injury demonstrated that low-dose IL-2 administration reduced allodynia, suggesting a Treg-mediated analgesic effect. Similarly, TNFR2 agonists were shown to reduce neuronal injury, attenuate peripheral and central inflammation, and promote reparative immune phenotypes within the central nervous system. These findings support the role of immune regulation in limiting pain chronicity.
P2X7 receptor and neuropathic pain
The P2X7 receptor (P2X7R), an ATP-gated ion channel expressed on immune cells, plays an important role in inflammatory and neuropathic pain mechanisms. Activation of P2X7R promotes inflammatory signaling and cytokine release, particularly IL-1β. Increased expression of P2X7R and elevated IL-1β levels have been observed in patients with neuropathic pain, suggesting a contribution of this pathway to chronic pain pathophysiology. These findings further reinforce the importance of neuroimmune signaling in pain sensitization and persistence.
Analgesia-permissive microenvironment
Beyond direct nociceptor modulation, immune cells may contribute to pain resolution through their interactions with other immune cells and by shaping the tissue microenvironment. Certain immune phenotypes promote anti-inflammatory and reparative processes, creating an analgesia-permissive environment that facilitates recovery and limits chronic sensitization. The dynamic interplay between immune cells and sensory neurons, therefore, appears fundamental in both the amplification and resolution of pain.
Questions and thoughts
Understanding the underlying pathophysiological mechanisms of pain is essential for broadening physiotherapists’ perspectives on pain management. According to the Pain Disability and Driver Model, clinicians’ understanding of nociceptive and neuropathic mechanisms is important for guiding both prognosis and treatment orientation. Further clinical tools that may assist physiotherapists in characterising nociceptive and neuropathic pain drivers will be discussed in the “Talk nerdy to me” section, exploring how they could relate to the immune control of pain proposed in this review.
Biologically grounded models are based on a mechanistic view of the human organism, in which modifying physiological pathways is assumed to reduce pain and improve function. While this perspective has contributed significantly to pain science, it may remain insufficient to fully account for the complexity of chronic pain. Pain is an embodied and subjective experience emerging from a complex, dynamic system in which biological, psychological, and contextual factors interact and cannot be meaningfully reduced to isolated subsystems.
Phenomenology in health sciences contributes to this broader understanding by focusing on the lived experience of the individual. As defined by the authors cited: “Phenomenology is a philosophical current which intends to observe and describe the meaning attributed to an experience from the consciousness of the person who is living it” (https://doi.org/10.3917/rsi.081.0021). Within this framework, physiotherapy practice is particularly well-suited to adopt an integrative and patient-centered approach. The iterative nature of clinical encounters allows for progressive exploration of the patient’s experience and the co-construction of a meaningful therapeutic strategy.
In this context, the physiotherapist is not merely a technician aiming to “fix” a dysfunction, but rather a clinical partner engaged in a collaborative process with the patient, moving away from a vertical model of care toward a shared and relational therapeutic alliance.
Talk nerdy to me
This section discusses currently available clinical tools that may help clinicians better understand the potential contribution of immune control of pain. None of these assessments has been validated to directly evaluate the specific status or precise contribution of neuroimmune drivers to pain. Rather, this section aims to bridge theoretical neuroimmune concepts with clinical practice by exploring how certain clinical findings may reflect the functional consequences of altered immune control of pain.
Conditioned pain modulation
Conditioned pain modulation (CPM) evaluates endogenous descending inhibitory pathways through the “pain inhibits pain” phenomenon. Impaired CPM may indicate deficient descending inhibitory control and altered neuroimmune regulation. From a theoretical perspective, such findings may reflect reduced anti-inflammatory cytokine activity (e.g., IL-10 and TGF-β), impaired endogenous opioid-mediated analgesia, altered regulatory T-cell activity, and persistent microglial activation contributing to central sensitization.
Sensory testing
Simple sensory testing, including light touch, pinprick, and thermal stimulation, may reveal allodynia or hyperalgesia. These findings could indicate peripheral and/or central sensitization processes. Mechanistically, inflammatory mediators, including IL-1β, IL-6, TNF-α, prostaglandins, histamine, NGF, and chemokines, lower nociceptor activation thresholds and increase neuronal excitability. In parallel, immune control of pain mechanisms involving microglial activation, BDNF release, ATP-P2X4/P2X7 signaling, and altered dorsal horn processing may contribute to amplified sensory responses and allodynia.
Pressure pain thresholds
Pressure pain threshold (PPT) testing evaluates the minimum pressure required to evoke pain and may provide indirect information regarding nociceptor sensitization and central pain amplification. Local reductions in PPT may reflect peripheral sensitization mediated by inflammatory cytokines, neuropeptides, prostaglandins, and NGF acting on mechanosensitive nociceptors. More widespread reductions in PPT, particularly at remote non-symptomatic sites, may suggest central sensitization and altered nociceptive gain within the central nervous system.
From a neuroimmune perspective, diffuse mechanical hypersensitivity may theoretically reflect persistent microglial activation, cytokine-mediated modulation of dorsal horn neurons, impaired descending inhibitory pathways, and sustained neuroimmune signaling involving IL-1β, TNF-α, IL-17, and ATP signaling.
Temporal summation testing
Temporal summation testing evaluates the progressive increase in pain perception following repetitive identical stimuli and reflects spinal cord excitability and pain facilitation mechanisms. Enhanced temporal summation is considered a clinical correlate of “wind-up” phenomena occurring at the dorsal horn level.
Theoretically, facilitated temporal summation may reflect persistent nociceptive input combined with neuroimmune-driven amplification of spinal neuronal excitability. Proposed contributors include microglial activation, ATP-P2X4/P2X7 signaling, BDNF release, cytokine-mediated modulation of synaptic transmission, and reduced inhibitory interneuron function. Elevated IL-1β, TNF-α, IL-6, and glial-derived mediators may contribute to increased synaptic responsiveness and sustained central sensitization.
Self-administered questionnaires
Self-administered questionnaires such as the CSI may indirectly reflect neuroimmune-related pain mechanisms by assessing symptoms such as widespread pain and sensory hypersensitivity, which could be associated with altered central processing, including microglial activity. However, as a self-reported tool, it remains subject to reporting bias. It nevertheless provides a useful entry point to explore the impact of pain on function, activity, and daily life.
Take-home messages
- Pain is not exclusively a neuronal phenomenon; it emerges from continuous interactions between nociceptors, immune cells, and central nervous system networks, forming a dynamic neuroimmune system.
- Immune cells are active regulators of pain, contributing not only to sensitization (e.g., cytokines, NGF, ATP signaling) but also to resolution through anti-inflammatory and pro-repair mediators (e.g., IL-10, IL-4, endogenous opioids, Tregs).
- Central sensitization involves neuroimmune amplification within spinal and supraspinal circuits, notably through microglial activation and cytokine/BDNF signaling.
- Neuroimmune communication is reciprocal: immune mediators influence nociceptors, while neurons actively modulate immune responses through neuropeptides and chemokines.
- Clinical tools such as QST, PPT, temporal summation, CPM, and questionnaires may not measure neuroimmune activity directly but can reflect its functional consequences at the systems level.
- Physiotherapy assessment can therefore benefit from a mechanism-informed interpretation of pain phenotypes, integrating sensory testing with biopsychosocial and immune control of pain-informed reasoning.
- Ultimately, pain should be understood as a complex, adaptive, and multidimensional experience requiring both mechanistic insight and patient-centered clinical integration.
Reference
How Nutrition Can Be a Crucial Factor for Central Sensitisation - Video Lecture
Watch this FREE video lecture on Nutrition & Central Sensitisation by Europe’s #1 chronic pain researcher Jo Nijs. Which food patients should avoid will probably surprise you!
